|
Контрольная работа: Процеси гідрування і дегідруванняНезалежно від вибору каталізатора та інших умов на селективність гідрування і дегідрування впливає температура. Звичайно, чим нижче температура, тим селективніше можна провести процес по більш реакційно-здатних групах або зупинити його на визначеній проміжній стадії. Небажані побічні процеси гідрогінолізу, крекінгу, дегідроконденсації мають більш високу енергію активації, чим дегідрування або гідрування. Так, для крекінгу н-бутану енергія активації приблизно дорівнює 250 кДж/міль, а для його дегідрування в н-бутен – 168-184 кДж/моль, що дозволяє підвищити селективність шляхом зниження температури. Оскільки при зменшенні температури одночасно зменшується швидкість процесу і продуктивність реактору, то практично в кожнім випадку можна знайти область оптимальних температур, що відповідає мінімуму економічних витрат. Додаткові обмеження на вибір цього оптимуму накладає оборотність реакції гідрування-дегідрування. Селективність залежить від часу контакту, що визначає фактичний ступінь конверсії вихідної речовини. Чим вона ближче до рівноважної, тим значніше розвиток послідовних реакцій більш глибокого гідрування, гідрогенолізу, крекінгу, що ведуть до зниження селективності. Тому для кожного процесу гідрування і дегідрування маються оптимальні ступінь конверсії і час контакту. Звичайне гідрування проводять при високому ступені конверсії (більш 90 %), а час контакту у різних випадках змінюється від часток хвилини до декількох годин. При більш високотемпературному дегідруванні, ускладненому оборотністю реакцій, ступінь конверсії складає іноді 20-40 %, а час контакту знаходиться в межах від часток секунди до декількох секунд. 6. Дегідрування парафінів і олефінів. Виробництво бутадієну та ізопрену Дегідрування парафінів, олефінів, а саме н-бутана, н-бутенов, изопентана і изопентанов має практичне значення у виробництві основних мономерів для синтетичного каучуку – бутадієну-1,3 і ізопрени. Бутадієн-1,3 (дивініл) СН2=СН-СН=СН2 безбарвний газ, який дає при змішанні з повітрям вибухонебезпечні суміші (2,0-11,5 %), який володіє деякою токсичністю, подразнюючи слизові оболонки і роблячи наркотичну дію. Він є основним мономером для синтетичного каучуку. Ізопрен (2-метилбутадієн-1,3) СН2=С(СН3)-СН=СН2 – безбарвна рідина, що утворюює при змішанні з повітрям вибухонебезпечні суміші (1,7-11,5 %) і є основною структурною одиницею натурального каучуку. Для одержання синтетичного каучуку ізопрен більш коштовний, чим бутадієн. Ізобутен – застосовується для одержання поліізобутена (вістанекс, опанол). Застосовується як добавка до мастил, для просочення тканин і як електроізоляційний матеріал. Полімеризацією изобутена з добавкою 2 % ізопрену одержують бутилкаучук, що відрізняється низькою газопроникністю і високою хімічною стійкістю. Вищі н-олефіни – мають велике значення для виробництва ПАР. 6.1 Дегідрування парафінів у моноолефіниДегідруванням олефінів одержують изобутен і вищі олефіни. При виробництві бутадієну-1,3 та ізопрену ця реакція є першою стадією двохстадійного процесу дегідрування. Дегідрування парафінів у моноолефіни в термодинамічному відношенні більш сприятливо, чим дегідрування алкілароматчних вуглеводнів і олефінів. Тому при припустимій температурі (6000С) не потрібно знижувати парціальний тиск і процес ведуть без розведення, при тиску лише небагато перевищуючому атмосферний. При дегидрировании парафинов С4 і С5 утвориться суміш ізомерних олефінів, наприклад, з бутану виходить бутен-1, цис- і транс-бутен-2:
ï СН3-С=С-СН3 і СН3-С=С-СН3 ï ï ê Н Н Н Крім того, виходять відповідні дієны, але в невеликій кількості, тому що умови для їх утворення несприятливі. Побічно протікає крекінг, ізомеризація, коксоутворення. У відношенні реакцій розщеплення парафіни більш реакційноздібні, ніж олефіни, тому нижчих вуглеводнів (СН4, С2Н4, С2Н6 та ін.) утвориться більше. Вважається, що ізомеризація в основному відбувається з олефінами, причому ізомерні олефіни (изобутен або н-пентен) частково гідруються. У продуктах реакції тому знаходяться ще ізомерні парафіни (ізобутан і н-пентен), які утворюють значну кількість коксу за рахунок реакцій ущільнення олефінів і дієнів, розкладання вуглеводнів на вуглець і водень:
CnH2n+2
продукти ізомери крекінгу олефінів парафінов Каталізатори повинні бути активні у відношенні основної реакції, але по-можливості не прискорювати процеси крекінгу, ізомеризації і закоксовування. Кращими є оксидноалюмохромові каталізатори на основі оксиду алюмінію (Al2O3), що містять 10-40 % Cr2O3 і 2-10 % оксидів лужних металів (Na2O, K2O, Be); останні служать для нейтралізації кислотних центрів оксиду алюмінію, що викликають крекінг та ізомеризацію. Ці каталізатори дуже чутливі до вологи і тому вихідні фракції С4 і С5 не повинні містити більш 1 мг водяних пар у 1 м3. Алюмохромові каталізатори активні до дегідрування н-бутану та ізопентану при температурі 500-6500С, підвищення температури веде до посиленого розвитку побічних реакцій, що мають більш високу енергію активації. Оптимальною вважають температуру 560-5900С – при дегідруванні н-бутану та 520-5600С – при дегідруванні найбільш активного ізопентану. Селективність падає при підвищенні ступеня конверсії вихідного парафіну (головним чином через уповільнення дегідрування при наближенні до рівноваги), тому її обмежують величиною 40-45 %. У зазначених умовах селективність процесу по н-бутену складає приблизно 75 %, по ізопентану – 70 %. Однак, алюмохромові каталізатори швидко закоксовуються і потрібне періодичне випалювання з них коксу повітрям при температурі 600-6500С. Через високу ендотермічність процесу під час відсутності розріджувача-теплоносія спочатку застосовували трубчасті реактори, що обігріваються топковими газами, чергуючи періоди дегідрування парафинов і регенерації каталізатора. Тепер використовуються системи з псевдозрідженим мікросферичним каталізатором. У них скомбіновані регенеративний принцип використання тепла і безперервна регенерація каталізатора (схема флюїд-реактора). Каталізатор виходить з реактора дезактивованим і надходить у регенератор, де повітрям випалюється кокс. За рахунок екзотермічності останньої реакції каталізатор розігрівається і знову надходить у реактор, де виконує додаткову роль теплоносія, що компенсує витрати тепла на ендотермічну реакцію дегідрування. У реакторах із псевдозрідженим каталізатором відбувається значне перемішування реакційної суміші, а це знижує продуктивність і селективність. Тому реактор постачають горизонтальними тарілками провального типу, що значно поліпшує показники процесу. Регенерований каталізатор надають на верхні розподільні ґрати, і псевдозріджений шар каталізатору і реакційні гази рухаються протитоком друг до друга, що створює найбільш сприятливий режим процесу (більш гарячий каталізатор контактує з частково прореагувавшою сумішшю і навпаки, чим досягається вирівнювання швидкостей реакції по всьому об’єму). У верхній частині реактора мається «гартівний» змійовик, де реакційні гази прохолоджуються н-бутаном, який йде на дегідрування. Завдяки цьому температура газів швидко знижується до 450-5000С и запобігається їх подальше розкладання. Регенерацію закоксованого каталізатору здійснюють також у псевдозрідженому шарі при протитоці газу та окиснювача, що надходить під нижні розподільні ґрати регенератору. Оскільки необхідно уникнути перегрівів, що ведуть до дезактивації каталізатора, регенерацію проводять сумішшю повітря з газами згорання палива, що містить 2-3 % (об.) кисню. При цьому оксид хрому все-таки частково окиснюється в CrО3 і, при відновленні останнього в реакторі, утворюється вода, що шкідливо впливає на властивості каталізатору. Щоб уникнути цього в десорбер регенератора надають топковий газ, що відновлює каталізатор, і ще нижче - азот, отдуваючий пари води і газу згорання. Після цього регенерований каталізатор при температурі 640-6500С підхоплюється транспортуючим газом і повертається до реактору. Технологічний процес дегідрування парафінів у відповідні олефіни складає три основні стадії: 1.Дегідрування парафінів з регенерацією каталізатора. 2.Виділення бутан-бутенової (або пентан-пентенової) фракції з продуктів реакції. 3.Поділ бутан-бутенової (або пентан-пентенової) фракції з одержанням бутенов (або ізопентенов). 6.2 Дегідрування вищих н-парафінів в олефины С12-С18Дегідрування вищих н-парафінов в олефины С12-С18 сильно відрізняється за своєю технологією від розглянутого вище процесу. Алюмохромові каталізатори виявилися непридатними, і для проведення процесу були розроблені платинові каталізатори з добавками металів і лугів, нанесених на оксид алюмінію, цеоліти або силікагель. Інша відмінність складається в необхідності розведення суміші воднем, що запобігає швидке закоксовування каталізатора і розвитку послідовних реакцій дегідрування. Мольне відношення водню і н-парафінів складає (6¸8):1 і загальний тиск 0,2-0,4 МПа, що несприятливо позначається на рівноважному ступені конверсії. Тому практичний ступінь конверсії досягає лише 11-14 % при селективності рівної 89-93 %. Дегідрування проводять у реакторі із суцільним стаціонарним шаром каталізатору в адіабатичних умовах при температурі 460-5000С. Після відділення водню і продуктів крекінгу каталізат переробляють двома способами: 1.Направляють на алкілування бензолу з наступним виробництвом сульфонолу і поверненням неперетвореного парафіну на дегідрування. 2.Розділяють його на н-парафіни і н-олефіни за допомогою молекулярних сит, використовуючи н-олефіни для інших синтезів. Одержання вищих н-олефінів з різним положенням подвійних зв'язків методом дегідрування виявилося економічно більш вигідним, чим термічним крекінгом парафинов. 6.3 Дегідрування олефінів Дегідрування олефінів у термодинамічному відношенні настільки ж несприятливо, як і дегідрування алкілароматичних вуглеводнів. Тому і тут для підвищення рівноважного ступеня конверсії при допутимій температурі (6000С) приходиться розбавляти реагуючу суміш водяною парою. Н-бутени або н-ізопентени, що надходять на дегідрування, незалежно від їх походження (із продуктів піролизу, крекінгу або дегідрування відповідних парафінів) являють собою суміш ізомерів, дегідруються тільки a-олефіни: СН2=СН-СН2-СН3 Для селективності ізомерів необхідна попередня ізомеризація з переміщенням подвійного зв'язку або утворенням поверхневого радикалу з делокалізованими електронами:
[CH3-CHLCHLCH2]· Крім цих цільових реакцій при дегідруваннІ протікають побічні процеси крекінгу, скелетної ізомеризації і коксоутворення. У результаті крекінгу з олефінів виходять метан і вуглеводні С2 і С3. Ізомеризація н-бутену веде до утворення ізобутена, але ця реакція особливо небажана для ізопентенів, коли отриманні пентени можуть далі дегідрувати у пентадієн-1,3 (пиперілен), а останній здатний заміняти цикл з утворенням циклопентадієну: (СН3)2С=СН-СН3 « СН3-СН2-СН=СН-СН3 «
« СН2=СН-СН=СН-СН3 ® Негативна роль останніх реакцій складається в одержанні домішок, які утрудняють очищення і виділення цільових речовин. При дегідруванні олефінів утворюються продукти ущільнення, кокс. Вважається, що головним їх джерелом є дієни, що схильні до реакцій конденсації з утворенням циклічних систем (дієновий синтез з наступної дегідроконденсацією ароматичних з'єднань). Нарешті, при дегідруванні олефінів за рахунок утворившегося водню виходить невелика кількість парафінів, що крекуються легше, ніж відповідні олефіни. Частина вуглеводнів і коксу піддається також конверсії водяною парою, унаслідок чого в газі утримуються оксиди вуглецю. парафін « олефіни « дієн « продукти ущільнення і кокс
низчи вуглеводні ізомеролефіни ізомери дієну Отже, маються рівнобіжні і послідовні шляхи утворення побічних процесів, селективність росте при зниженні двох факторів: температури (через більш високу енергію активації побічних реакцій) і ступеня конверсії; вибір цих величин обумовлений економічними розуміннями. Тому каталізатори повинні прискорити переважно дегідрування та ізомеризацію з переміщенням подвійного зв'язку, не повинні бути малоактивними у відношенні крекінгу, скелетної ізомеризації і коксоутворення. Кращими є кальцийнікельфосфатні каталізатори ИМ-2204 (Са8Ni(PO4)6), що містять промотируючу добавку 2 % Cr2O3, їх випускають у формованном виді для роботи в стаціонарному шарі. Характерна риса – швидке закоксовування і втрата реактивності, тому потрібне часте випалювання коксу. Періоди дегідрування і регенерації чергуються кожні 5 хвилин, попередньо продуваючи реактор водяною парою. Дегідрування ведуть, розбавляючи вихідну суміш водяною парою в об'ємному співвідношенні 20:1, при об'ємній швидкості по газоподібному вуглеці 150-200 ч-1 і загальному тиску тільки небагато перевищуючому атмосферний (щоб перебороти гідравлічний опір шару каталізатора і наступної апаратури); температура складає 600-6500С, ступінь конверсії – 40-45 %, селективність – 85 % - при дегідруванні н-бутенів. Для ізопентану, більш реакційноздібного і більш схильного до побічних реакцій, температура складає 550-6000С, ступінь конверсії – 40 %, селективність – 84 %. Для проведення процесу використовують реактори зі стаціонарним шаром каталізатора, що не мають поверхонь теплообміну. При цьому пара відіграє роль теплоносія, що не дозволяє суміші надмірно охолоджуватися (перепад температур між входом і виходом складає 30-400С). Крім кальцийнікельфосфатних контактів застосовують залізооксидні каталізатори (К-16), що містять 25-90 % Fe2O3, 2-50 % Cr2O3, до 15 % К2СО3 та інші компоненти. Вони є саморегенеруючими і здатні працювати до 24 годин, після чого регенеруються. На цих каталізаторах процес ведуть при розведенні вихідної суміші водяною парою в об'ємному співвідношенні 10:1, ступінь конверсії складає 17-20 %, селективність – 80-85 %. 6.4 Окиснювальне дегідрування олефінівДегідрування олефінів має істотні недоліки: циклічність роботи каталізатора і реакційного вузла, порівняно низькі ступінь конверсії і селективність, велику витрату енергії. Найбільш перспективний процес окисного дегідрування олефінов, коли в реакційну суміш уводять кисень або повітря, що зв'язує водень у воду: СН2=СН-СН2-СН3 + 0,5О2 Цим усувається оборотність реакції дегідрування і знімаються термодинамічні обмеження ступеня конверсії олефіна, а процес з адіабатичного перетворюється в екзотермічний, що робить його більш зручним для практичної реалізації. Крім того, як і в інших реакціях окиснення, каталізатор може служити довгий час без регенерації. Каталізаторами окисного дегідрування олефінів виявилися оксидні композиції: Bi+Mo, Bi+Mo+P, Bi+W, Fe+Sb та ін. Усі вони активні при температурі 400-6000С І працюють у по окиснювально-відновному механізму за участю кисню кристалічних ґрат: К-О- + С4Н8 « К-О-С4Н8 ® К + Н2О + С4Н6 2К + О2 ® 2К-О- В одному з варіантів процесу, щоб не піддавати олефіни дії молекулярного кисню, дегідрування олефіна окисним каталізатором і окиснення відновленого каталізатора проводять у двох різних реакторах із псевдозрідженим шаром каталізатора. В усіх випадках потрібно розбавляти олефін водяним пором в об'ємному відношенні від 1:5 до 30:1, ступінь конверсії складає 70-80 %, селективність – 90-95 %. 6.5 Одностадійне дегідрування парафінів у дієниДвохстадійний метод одержання бутадієну-1,3, що відрізняється порівняно високим виходом (до 65 %), має ряд недоліків: необхідність поділу газових сумішей після кожної стадії, підвищені енергетичні і капітальні витрати. З цієї причини проводилися інтенсивні роботи зі створення одностадійного процесу перетворення н-бутану та ізопентану в бутадієн-1,3 та ізопрен. При одностадійному процесі протікають дві оборотні послідовні стадії дегідрування: С4Н10 Рівноважний склад цієї системи залежить від температури і тиску. При підвищенні температури рівноважна концентрація н-бутану різко падає, вміст н-бутенів проходить через максимум при температурі 800 К, а кількість бутадієну росте, але не настільки значно, через одночасне утворення водню на обох стадіях. Це показує, що для одностадійного процесу варто вибирати більш високу температуру, чим на першій стадії дегідрування парафінів, і знижений парціальний тиск реагентів. Крім того, потрібен каталізатор, що відповідним чином прискорював би обидві реакції дегідрування, наприклад, алюмохромовий. Оскільки при роботі з цим каталізатором не можна використовувати водяну пару, то як розріджувач був розроблений процес, що йде за зниженим тиском (0,015-0,02 МПа) і температурі 580-6000С, що є середнім між оптимальними для першої і другої стадії дегідрування парафінів. Через застосування вакууму реактори з рухомим каталізатором виявилися непридатними для одностадійного процесу. Сильне відкладення коксу і необхідність частої регенерації обумовили використання регенеративної системи Гудрі. Реакційний вузол при одностадійному процесі включає ряд блоків, що складаються з 5-8 горизонтальних реакторів зі стаціонарним шаром каталізатору. Кожен реактор працює періодично, за регенеративним принципом використання тепла. У період випалювання коксу і регенерації каталізатору останній розігрівається до температури 6000С. Потім йде евакуація газів згорання за допомогою вакууму (1,5-2 хв.) і дегідрування, коли тепло насадки використовується для проведення ендотермічного процесу, і вона прохолоджується до мінімально припустимої температури (5800С). Після цього реактор продувають перегрітою водяною парою для витиснення вуглеводнів (1,5-2 хв.) і знову проводять регенерацію каталізатору. Щоб охолодження в період дегідрування відбувалося не занадто швидко, до каталізатора додають гранули прожареного глинозему, що грає роль акумулятора тепла, але й у цьому випадку стадії дегідрування і регенерації тривають усього по 5-9 хв., із загальною тривалістю циклу роботи реактора 15-20 хв. Усі переключення потоків проводяться автоматично і завдяки наявності у блоці 5-8 реакторів створюється безперервний постійний потік вихідних речовин і одержуваних продуктів. Ступінь конверсії вихідної сировини становить 20-30 % при селективності 55 %. Контактний газ містить приблизно 11 % бутадієну-1,3 і 25-30 % бутенів. Через проведення обох стадій дегідрування не в оптимальних для них умовах селективність менше, ніж при двохстадійному процесі, але це компенсується зниженням капітальних вкладень і енергетичних витрат завдяки скороченню деяких стадій виробництв. 7. Хімія і технологія процесів гідрування У промисловості основного органічного синтезу широко використовуються процеси гідрування вуглеводнів, кисень-, азотутримуючих з'єднань з метою одержання насичених вуглеводнів, кетонів, карбонових кислот, спиртів, амінів. 7.1 Гідрування вуглеводнів7.1.1 Гідрування по С=С зв'язкуЯк каталізатор для гідрування олефінів по С=С зв'язку використовували платинову чернь, потім стали використовувати платину, скелетний нікелевий каталізатор (нікель Ренея), нікель на носіях, мідь, змішані каталізатори (мідь, хромоксидний, цинк і хромоксидний) і інші каталізатори. Найбільш типові металевий нікель і нікель, обложений на оксиді алюмінію, оксиді хрому та інших носіях. У їх присутності висока швидкість реакції досягається при температурі 100-2000С і тиску 1-2 МПа. Якщо вихідна сировина містить сіркоутворюючи з'єднання, рекомендується застосовувати каталізатори стійкі до отруєння сіркою (сульфіди нікелю, вольфраму, молібдену), на цих каталізаторах швидкість досягається при температурі 300-3200С і тиску 25-30 МПа. Гідрування по С=С зв'язку протікає дуже легко і майже з теоретичним виходом. При цьому реакційна здатність олефінів залежить від ступеня заміщення атомів вуглецю, що знаходяться при подвійному зв'язку. Швидше всіх гідрується етилен, а для його гомологів швидкість реакції внаслідок екстрагуючого впливу заступників падає в ряді: СН2=СН2 > RCH=CH2 > RCH=CHR > R2C=CH2 > R2C=CHR > R2C=CR2 Дієнові вуглеводні з прямим ланцюгом гідруються швидше олефінів. У залежності від будівлі дієну водень може приєднуватися на початку в 1,4-положення або одночасно у 1,4 і 1,2-положення, причому часто вдається зупинити реакцію на цій стадії. Більш глибоке гідрування приводить до утворення парафінів: ô ô ô ô
ô ô ô ô ô ô ô ô ô ô ô С=С-С=С ô ô ô ô ô ô ô ô СН-СН-С=С ô ô Аналогічним чином гідруються полієнові вуглеводні, що поглинають два атоми водню на кожен подвійний зв'язок, аж до утворення цілком гідрованих з'єднань. При відповідному дозуванні водню і виборі селективного каталізатору вдається провести чисте гідрування таких з'єднань. У циклоолефінах подвійний зв'язок здатний приєднувати водень у присутності тих же каталізаторів гідрування. Наявність алкільних заступників при подвійному зв'язку також зменшує швидкість реакції. При гідруванні циклоолефінів необхідно дотримувати запобіжного заходу, тому що можуть одержати значний розвиток побічні реакції гідрогенолізу по вуглець-вуглецевому зв'язку, що при гідруванні олефінів з відкритим ланцюгом не грають істотної ролі. У більш жорстких умовах відбувається розкриття циклу при дії водню особливо у три-чотиричленних циклічних системах. Вуглеводні із шістьма і більш вуглецевими атомами в циклі в умовах гідрування можуть також ізомерізуватися в більш стабільні п'ять- і шестичленні цикли:
С8Н18 Ці побічні реакції обмежують вибір температури при гідруванні олефінів і особливо циклоолефінів, це необхідно мати на увазі при визначенні оптимальних умов. Гідрування вуглеводнів по С=С зв’язку широко застосовується при стабілізації крекінг-бензину, при селективному очищенні рідких продуктів піролізу від олефінів і т.д. В основах органічного синтезу цей тип реакцій гідрування використовується для одержання деяких циклоолефінів (циклоалкенів) і циклопарафінів (циклоалканів). Циклопентен, циклооктен, циклододілен є коштовними полімерами для одержання синтетичного каучуку, а відповідно циклопарафіни застосовуються для одержання дикарбонових кислот, циклоалканів і лактамів. Для цього синтезу проводять гідрування циклопентадієну (виділенного з продуктів піролізу) циклічних дімерів і тримерів бутадієну. Гідрування до циклоолефінів можна здійснити за допомогою гетерогенних каталізаторів (нікель, кобальт на носіях); у випадку синтезу циклооктану і циклододенану кращі результати отримані з гомогенними каталізаторами. Останні особливо придатні для селективного гідрування поліолефінів у моноолефіни – циклопентен, циклооктен, циклододецен. З цих каталізаторів найбільш розповсюджений гідрокарбоніл родію з трибутилфосфориновим лігандом НRh(CO3)×P(C4H9)3. У його присутності при температурі 1400С і тиску приблизно рівному 2,5 МПа досягається ступінь перетворення циклододекатрієну близько до 100 % і селективність по моноолефіну понад 95 %.
циклопентен циклооктен
циклододілен 7.1.2 Гідрування по СºС зв'язкуАцетилен і його гомологи гідруються повільніше олефінів, але проміжно утворені олефіни легко витісняються з поверхні каталізатора через меншу здатність до сорбції і тому можуть бути отримані як цільові продукти. Селективне гідрування до олефінів здійсненно при каталізі платини і паладия на носіях, а також молибдатами кобальту, нікелю, заліза та ін. При більшому часі контакту гідрування йде до утворення парафіну: СНºСН З гомологів ацетилену легше реагують з воднем ті, які мають потрійний зв'язок на кінці ланцюга. З'єднання з потрійним зв'язком у середині ланцюга гидруються повільніше, і в цьому випадку перша і друга стадія розділені більш чітко, чим у випадку ацетилену. При каталізі нікелю, а особливо міді, і при недоліку водню з ацетилену виходять інші продукти, що представляють собою суміші його частково гідрованих полімерів. На мідному контакті утвориться тверда полімерна речовина – купрен. Він застосовується в якості наповнювача при виготовленні різних матеріалів. Ацетилен можна використовувати для одержання етилену при температурі 180-3200С і 1,5-2,0-кратному надлишку водню з паладієвим каталізатором на силікагелі. Подібний процес застосовують для селективного очищення етилену від домішки ацетилену (останній завжди утвориться при піролизі вуглеводневих газів, при якому виділяється також водень). Гідроочищення від ацетилену досягається пропущенням газу через контактний апарат з каталізатором (паладій; нікель на носії; молібдати нікелю і кобальту). 7.2 Гідрування ароматичних вуглеводнівКаталізаторами гідрування ароматичних систем можуть служити всі метали VIII групи, але в промисловості застосовують, головним чином, нікель на носії, а особливо Сr2О3. З таким контактом достатня швидкість процесу досягається при температурі 120-2400С, при цьому, на відміну від гідрування олефінів, необхіден підвищений тиск (1-5 МПа) не тільки для прискорення реакції, але і для збільшення рівноважного ступеня конверсії, тому що термодинамічне відношення в цьому випадку менш сприятливе для гідрування. Ароматичні вуглеводні потрібно попередньо очищати від з'єднань сірки. Порівняльна стабільність ароматичної системи обумовлює її меншу реакційну здатність при гідруванні в порівнянні з олефінами. Так відносні швидкості гідрування бензолу, циклоолефіну і подвійних зв'язків у бічному ланцюзі арилолефіну такі (в умовних одиницях):
¾ СН2=СН3 1 150 900 З цієї причини, незважаючи на послідовно-рівнобіжний тип реакції зі східчастим насиченням зв'язку, у реакційній масі не виявляються циклічні олефіни:
Однак у спеціальних умовах з бензолу можна одержати циклогексен (на гомогенних каталізаторах) або циклогексилбензол (на гетерогенних біфункциональних каталізаторах): С6Н6 Гомологи бензолу гідруються з меншою швидкістю, чим сам бензол - це явище дезактивуючого впливу заступників при гидруємих зв'язках. Вони гидруються в першу чергу при наявності в ароматичному вуглеводні ненасиченого бічного ланцюга. У присутності мідного каталізатора, некаталізуючого гідрування ароматичних систем, насичуються тільки бічні ланцюги, а на нікелі, при надлишку водню, відбувається повне гідрування, наприклад стиролу:
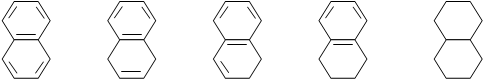
¾ С2Н5 Ароматичні вуглеводні з конденсованими ядрами звичайно гідруються східчасто, з поступовим насиченням зв'язків і кілець. Гідрування нафталіну йде через наступні стадії (із проміжною ізомеризацією aa' дегідронафталіну в більш стабільний a-, b-ізомер):
тетралін декалін Умови гідрування ароматичних вуглеводнів багато в чому визначаються побічними реакціями гідрогенолізу з розщепленням бічних ланцюгів і циклів. Так, з бензолу на нікелевому каталізаторі при температурі 2900С утвориться метан (до 71 %) і вільний вуглець:
Ще легше розщеплюються бічні ланцюги, особливо більш довгі. Наприклад, з н-пропілбензолу при гідруванні можна одержати не тільки пропілциклогексан, але також этил-метилзамісні, циклогексан і нижчі парафіни. Ці побічні реакції визначають верхню межу температури при гідруванні ароматичних вуглеводнів. У промисловості процеси гідрування ароматичних вуглеводнів застосовують для синтезу ряду важливих продуктів, особливо циклогексану, що застосовується для одержання циклогексанолу, капролактаму, адипінової кислоти. 8. Технологія газофазного гідрування Гідрування в газовій фазі здійснюють, пропускаючи суміш водню з парами органічної речовини через гетерогенний контакт. Цей процес застосовують для речовин, летючість яких при температурі реакції достатня для створення необхідного парціального тиску їх у парогазовой суміші. При великому надлишку водню, високій температурі або зниженні загального тиску – цим шляхом можна гидрувати і менш летучі речовини. Процес широко використовують для гідрування бензолу, фенолу, нітробензолу, аліфатичних альдегідів, кетонів і т.д. Переважна більшість реакцій газофазового гідрування проводиться зі стаціонарним каталізатором у виді кульок, таблеток або в інших формах, розміром 3-5 мм. Тільки в рідких випадках застосовують дрібнодиспергований контакт – для процесу з псевдозрідженим каталізатором. 8.1 Типи реакційних пристроївЧерез низькі коефіцієнти тепловіддачі від газу до стінки проблема тепловідводу при газофазном гідруванні значно складніша, ніж при рідинофазном. Вона ще більш ускладнюється при нерухомому шарі каталізатора, зерна якого перешкоджають дифузії реагентів та їхньому охолодженню. У залежності від ступеня экзотермічності реакції відвід тепла досягається трьома основними способами, що визначають конструктивні особливості реакторів гідрування. У трубчастих апаратах, які застосовуюються для проведення сильно екзотермічних процесів гідрування (відновлення нітроз’єднань, гідрування ароматичних з'єднань), каталізатор поміщають у трубах діаметром 25х50 мм. Парогазовую суміш водню з органічним реагентом звичайно надають зверху (іноді знизу) і реакція протікає в трубах на зернах каталізатора. Виділене тепло знімається хладоагентом, який циркулює в міжтрубному просторі. У якості хладоагенту особливо підходить киплячий водний конденсат, у цьому випадку можна утилізувати тепло реакції для одержання водяної пари. Ступінь використання реакційного об’єму у цих апаратах невеликий, тому для менш екзотермічних реакцій застосовують апарати із суцільним шаром каталізатору, що поміщається на дирчастих полках або в спеціальних кошиках у кілька шарів. У просторі між шарами маються холодильники, але з верхньою подачею парогазової суміші. Іноді використовують трохи адіабатичних реакторів із суцільним шаром каталізатору. Для ще менш экзотермічних реакцій (гідрування насичених альдегідів) можна обмежитися подачею між шарами каталізатору холодного водню, який сприймає надлишкове тепло. При газофазном гідруванні карбонових кислот або їх ефірів у спирти можна взагалі обійтися без охолодження реакційної суміші. Для роботи за високим тиском використовуються апарати, подібні застосовуваним при синтезі аміаку. Каталізатор розміщають у кілька шарів у спеціальній катализаторній коробці, яку монтують поза реактором і яку можна виймати з нього і вставляти при заміні каталізатору. У кільцевий простір між корпусом реактору і каталізаторною коробкою надають холодний водень або реакційну суміш для часткового відводу тепла і запобігання корпусу від дії високих температур. У кілька місць по висоті коробки вводять холодний водень, щоб не послабляти корпус реактора, усі труби виведені не збоку, а через масивну кришку у днищі. При проході парогазової суміші через суцільний шар каталізатору температура трохи підвищується, тому висоту шару потрібно підбирати так, щоб не відбувалося надмірного перегріву і температура знаходилася у припустимих оптимальних межах. Важливим методом регулювання температури при газофазному гідруванні є застосування великого надлишку водню в порівнянні з теоретично необхідним. Він складає в різних процесах від 5:1 до (20¸30):1. Надлишковий водень акумулює выділене тепло, запобігаючи надмірний перегрів реакційної маси. В кожному випадку мається оптимальний надлишок водню, при встановленні якого необхідно враховувати витрати на його рециркуляцію. Цікавим варіантом оформлення процесу є сполучення двох реакторів: реактору для гідрування в рідкій фазі із суспендированим каталізатором і реактору для гідрування в газовій фазі з стационарним каталізатором. Тут у першому апараті також здійснюється основна частка перетворень за тим або іншим способом відводу тепла. Компоненти суміші випаровуються в струмі водню і гідрування завершується в другому апараті, що не вимагає теплообмінних пристроїв. Ця схема особливо підходить для гідрування бензолу в циклогексан, коли досягається майже повна конверсія бензолу при високій продуктивності установки. 9. Технологія рідинофазного гідрування Рідинофазне гідрування проводять шляхом барботирування водню через рідку реакційну масу. Цим способом завжди гідрують висококиплячі речовини (жири, вищі карбонові кислоти та їх ефіри, динітрили, динітроз’єднання), оскільки для їхнього перекладу в стан насиченої пари був би потрібен надмірний надлишок водню і зайві економічні витрати. Однак за високим тиском в рідкій фазі можна гідрувати і більш летучі речовини. Процеси рідинофазного гідрування класифікують по декількох основних ознаках: 1. Гідрування в середовищі гідруємого з'єднання без сторонніх добавок. У цьому випадку низька реакційна маса складається з вихідної органічної речовини, у якій поступово накопичуються утворюємі продукти. Такий спосіб застосовується найбільш часто. 2. Гідрування в розчині речовин інертних в умовах реакції. Цей спосіб використовується при відновленні з'єднань, твердих при робочій температурі (одержання сорбітаманіїту з вуглеводів у водному розчині, гідрування полімерів) або схильних (при високій їх концентрації) до підвищеного утворення побічних продуктів. Так, альдегіди гідрують у виді їх розчинів у відповідних спиртах, щоб уникнути розвиток процесів альдольної конденсації. 3. Гідрування в емульсіях, наприклад відновлення ароматичних динітроз’єднань у водній емульсії. При цьому поліпшуються умови відводу реакційного тепла, полегшується виділення розчиненого у воді діаміну, запобігається розкладання термічно нестабільних динітроз’єднань. Залежно від форми застосовуваних каталізаторів рідинофазні процеси поділяються на три групи: 1. З тонкодиспергованим каталізатором, нерідко одержуваним безпосередньо в масі гідруємої речовини (розкладання форміатів або карбонілів металу). Такий каталізатор дуже активний, але його важко відокремлювати від гідрогенізату при наступній переробці. 2. Із суспендованим каталізатором у реакційній масі, який подрібнюється до визначеного розміру. Він легше відокремлюється від гідрогенізату при наступному фільтруванні, але поступово стирається при роботі, що веде до зносу стінок апарата. 3. З нерухомим (стаціонарним) каталізатором, який використовується у виді гранул різної форми, досить великих, щоб їх не несли потоки рідини і газу. Цей варіант найбільш удалий, тому що виключається стадія наступного фільтрування гідрогенізату. Рідинофазні процеси можна здійснювати як періодично, так і безупинно. 9.1 Типи реакційних пристроївРеакційна маса в процесах рідинофазного гідрування є, як правило, трифазною (рідкий реагент, твердий каталізатор, газоподібний водень). Реакція протікає на поверхні каталізатору, причому її швидкість, за інших рівних умов, залежить від концентрації водню в рідині, що залежить від тиску, швидкості розчинення водню в реакційній масі і швидкості його дифузії к поверхні каталізатору. Підвищенню швидкості сприяє високий тиск водню і перемішування реакційної маси, що є характерним для всіх процесів рідинофазного гідрування. Оформлення реакційного вузла для рідинофазного гідрування сильно залежить від экзотермічності реакції і способу відводу тепла. Тільки в рідких випадках виділення тепла настільки мало, що реакцію можна здійснити без охолодження (гідрування карбонових кислот та їх ефірів). У випадку гідрування летучих речовин (перетворення бензолу в циклогексан) іноді відводять тепло за рахунок випару компонентів суміші, які конденсують і повертають до реактору. Найбільш часто для процесів з диспергованими і суспендованими каталізаторами здійснюють примусове охолодження за допомогою внутрішніх або виносних холодильників; у цьому випадку тепло реакції може використовуватися для одержання пари. Тепловідвод утруднений для процесів зі стаціонарним каталізатором, тоді найчастіше ведуть східчасте охолодження суміші. Останнім часом для рідинофазного гідрування застосовують комбінування двох реакторів із суспендованим і стаціонарним каталізатором. В основному реакція протікає у першому реакторі, у якому тепло відводиться одним з описаних методів. В другому реакторі відбувається тільки невелика частка перетворень, причому охолодження не потрібно зовсім. Завдяки комбінуванню апаратів, близьких відповідно до моделей повного змішання, та ідеального витиснення досягаються висока продуктивність і ступінь перетворення сировини. У ряді випадків колони можна виконувати зі звичайної сталі. Якщо процес проводиться за високим тиском, який сприяє водневій корозії, або з агресивними речовинами (карбонові кислоти та ін.) потрібні спеціальні сталі або облицювання сталевого корпуса легованою сталлю та іншими корозійнно-стійкими металами. література1. Лебедев Н.Н. Химия и технология основного органического и нефтехимического синтеза. – М.: Химия, 1988. 2. Одбашен Г.В., Швец В.В. Лабораторный практикум по химии и технологии основного органического и нефтехимического синтеза. – М.: Химия, 1992. 3. Тимофеев В.С., Серафимов Л.А. Принципы технологии основного органического и нефтехимического синтеза. – М.: Химия, 1992. 4. Кирпичников П.А., Лиакумович А.Г., Победимский Д.Г., Попова Л.М. Химия и технология мономеров для синтетических каучуков. –Л.: Химия, 1981. 5. Черный И.Р. Производство сырья для нефтехимического синтеза. – М.: Химия, 1983. 6. Спарченко В.К. Дегидрирование углеводородов. – Киев: Наукова думка, 1981. |
|||||||||||||||||||||||
Страницы: 1, 2
 |
||
| НОВОСТИ |  |
|
|
||
| ВХОД | |
|
|
|||||
Рефераты бесплатно, реферат бесплатно, рефераты на тему, сочинения, курсовые работы, реферат, доклады, рефераты, рефераты скачать, курсовые, дипломы, научные работы и многое другое. |
||
При использовании материалов - ссылка на сайт обязательна. |
||

 СН2=СН-СН2-СН3
СН2=СН-СН2-СН3
 СН-С=С-СН
СН-С=С-СН цикло С6Н10(СН3)2
цикло С6Н10(СН3)2