|
Курсовая работа: Цитогенетическая и молекулярно-цитогенетическая характеристика микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди ДжорджиТаблица 3 Корреляция генотип—фенотип при синдроме Прадера-Вили
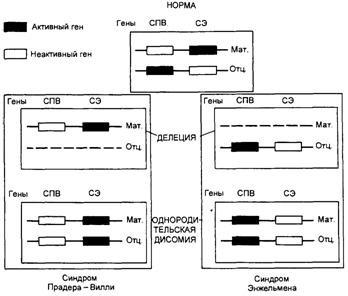
Наиболее частой причиной возникновения СПВ и СА является протяженная (до 4 млн. п.н.) деления критического района 15(q11—q13), которую находят у 70—75 % больных с этими синдромами. Делецию при СПВ обнаруживают на отцовской хромосоме 15, а при СЭ делеция той же области на ее материнском гомологе. Второй причиной возникновения СПВ и СЭ оказалась однородительская дисомия, т.е. наследование обоих гомологов хромосомы 15 от одного из родителей. С помощью ДНК-маркеров региона делеции путем блотт-гибридизации по Саузерну, а также анализа метилирования было продемонстрировано различное родительское происхождение хромосомы 15: в первом случае отцовское, во втором — материнское. Поскольку этот регион хромосомы 15 идентичен аналогичному региону хромосомы 2 мыши, для которого хорошо известен геномный импринтинг, исследования стали проводить в этом направлении. Оказалось, что регион СПВ активен на отцовской хромосоме (при наличии делеции на отцовской хромосоме или материнской ОРД отсутствуют отцовские гены) и не активен на материнской (рис. 3). Из рис. 1 видно, что при СПВ не экспрессируются отцовские гены, а при СЭ — материнские гены. Материнская ОРД наблюдается в 25 % случаев СПВ, а отцовская ОРД становится причиной возникновения СЭ в 3—5 % случаев [1]. В последние годы появились сообщения еще об одной причине развития этих синдромов у пациентов, у которых не было найдено ни типичных делеций, ни ОРД, но зато в семьях таких больных встречались повторные случаи синдрома. В ходе исследования в проксимальном участке хромосомы 15 были обнаружены противоположно импринтированные гены — кандидаты СПВ и СЭ, соответственно SNRPN и UBE3A, в которых выявили мутации.
Рис. 3 Механизмы возникновения синдромов Прадера—Вилли и Ангельмана через делецию участка хромосомы 15 или однородительскую дисомию по хромосоме 15. СПВ — синдром Прадера—Вилли; СА — синдром Ангельмана; мат. — материнская хромосома; отц. — отцовская хромосома Ген SNRPN кодирует полипептид N малого ядерного рибонуклеопротеина, активно экспрессируется исключительно на отцовской хромосоме 15 и репрессирован на материнском гомологе, т.е. мутации в этом гене вовлечены в патогенез СПВ. Критический регион для СЭ расположен дистальнее (локус D15S10), который экспрессируется только в материнских хромосомах. Предполагают, что мутации при СЭ есть в гене UBE3A кодирующем убиквитинлигазный белок ЗА. Экспрессия этого белка выявлена во всех тканях человека, причем в ряде структур мозга ген UВЕЗА активен лишь на материнской хромосоме. Дефицит материнской копии этого гена в клетках Пуркинье (грушевидных невроцитах мозжечка) и нейронах гиппокампа может, по-видимому, объяснить клиническую картину СЭ (умственная отсталость, атаксия, тремор и др.). Таким образом, в районе хромосомы 15(q11—q13) имеются близко расположенные, но противоположно импринтированные локусы, отвечающие за возникновение этих двух синдромов. Эта область хромосомы 15 чрезвычайно существенна для нормальной переустановки геномного импринтинга. Она названа центром импринтинга (IC). Мутации в данной области приводят к ошибкам импринтинга, т.е. теряется способность стирать отпечаток предшествующего поколения. Так, если в сперматогенезе отца не происходит замены «женского» импринта на «мужской» на его материнской хромосоме, то в следующем поколении возникнет состояние, аналогичное материнской ОРД, которое будет сопровождаться фенотипом СПВ. Нарушение установления «женского» эпигенотипа на отцовских хромосомах в овогенезе матери приведет к развитию СА у потомства. Повторный риск для трех групп семей при СПВ и СЭ будет существенно различаться. Так, при делециях он будет ниже 1 %, при ОРД риск также низкий, но в этом случае нужно учитывать возраст матери, который может увеличивать риск. При мутациях в центре импринтинга повторный риск будет существенно выше не только для родителей больного, но и ближайших родственников. Связь геномного импринтинга с другой наследственной патологией человека на уровне хромосом или отдельных генов также отчетливо прослеживается и в настоящее время широко изучается. Так, например, при хорее Гентингтона и спинно-мозжечковой атаксии I заболевание возникает раньше и протекает тяжелее, если унаследованные гены имеют отцовское происхождение. При нейрофиброматозе 1 и 2, миотонической дистрофии, наоборот, заболевание имеет более раннее начало и тяжелое течение при унаследовании мутантных генов от матери. Не вызывает сомнения причастность геномного импринтинга к этиологии опухолевого роста. Выключение импринтинга, а также потеря гетерозиготности или ОРД по хромосомам или их участкам, содержащим импринтированные локусы, могут приводить к функциональной нуллисомии генов-супрессоров опухолевого роста или к аберрантной экспрессии протоонкогенов, что может лежать в основе возникновения рака. Кроме того, вероятность ОРД повышается не только с возрастом матери, но и у носителей изохромосом, робертсоновских и реципрокных транслокаций. Следует иметь в виду, что ОРД (изодисомия) может привести к гомозиготизации определенных регионов хромосомы и быть причиной аутосомно-рецессивной патологии. Такие случаи описаны, например, при муковисцидозе. Точные механизмы, лежащие в основе дифференциальной экспрессии материнских и отцовских геномов, пока не известны. Основную роль в этом процессе отводят специфическому метилированию цитозиновых оснований ДНК. Важнейшими особенностями метилирования ДНК являются, во-первых, стабильное сохранение в ряду многих поколений клеток, а во-вторых, прямое или косвенное влияние на экспрессию генов. Специфическое для пола метилирование некоторых участков генома устанавливается во время гаметогенеза. Известно, что некоторые повторяющиеся и даже уникальные последовательности ДНК являются недометилированными в яйцеклетках и гиперметилированными в сперматозоидах. Такие различия между родительскими хромосомами сохраняются и после оплодотворения и стабильно передаются в следующие клеточные поколения. Как правило, активный ген ассоциируется со сниженным метилированием или его отсутствием, а неэкспрессирующий генетический регион — с гиперметилированием. Тканеспецифичное метилирование цитозиновых остатков ДНК осуществляется в ходе гамето- и эмбриогенеза с помощью ДНК-метилтрансфераз. Значительная доля импринтированных генов (до 15 %) ассоциирует с антисмысловыми транскриптами. Такие транскрипты представлены обычно антисмысловой РНК, происходящей из интронов некоторых генов, и колинеарной ДНК. Эта антисмысловая РНК не выполняет кодирующих функций и, возможно, является регуляторной. Предполагают, что существуют и другие механизмы, регулирующие дифференциальную активность отцовских и материнских генов. Описывают две модели смены эпигенотипа хромосом в гаметогенезе. Согласно первой, переключение эпигенотипа происходит только в той из гомологичных хромосом, которая унаследована от родителя противоположного пола, а вторую хромосому модификации не затрагивают. Вторая модель предполагает предварительное устранение («стирание») существующего эпигенотипа на обеих родительских хромосомах с последующим установлением импринта, соответствующего данному полу. За последние годы в результате многочисленных исследований метилирования и функционирования импринтированных генов в клетках зародышевого пути были получены убедительные доказательства в пользу второго предположения. Таким образом, хотя роль метилирования в обеспечении аллельспецифической экспрессии генов несомненна, остается неясным, является ли метилирование первичным эпигенетическим сигналом, который «стирается» и устанавливается в га-метогенезе, или представляет собой некий вторичный процесс по отношению к более ранней стадии импринтинга и служит лишь для поддержания ранее установленного импринта. Хотя в настоящее время не вызывает сомнения, что метилирование ДНК является эпигенетической меткой и оно достаточно хорошо изучено и характерно практически для всех импринтированных генов и локусов, нельзя исключить и другие пока еще неизвестные механизмы. Дальнейшее изучение геномного импринтинга (особенно в рамках функциональной геномики) будет иметь существенное значение для понимания тонких механизмов регуляции генной активности в онтогенезе и его связи с патологией человека [3]. Импринтинг генов ведет к необычным последствиям. У мужчин материнская копия хромосомы 15 содержит в себе знак того, что она пришла от матери. Но уже в следующем поколении у дочери или сына эта же хромосома будет содержать знак отцовского происхождения. В какой-то момент должно произойти переключение знака хромосомы на противоположный. Нет сомнений в том, что такое переключение происходит, поскольку только этим можно объяснить синдром Ангельмана. Никаких видимых повреждений на хромосоме 15 нет, просто две хромосомы ведут себя так, как будто обе произошли от отца. Это объясняется тем, что в нужный момент в организме матери не произошло переключение знака хромосомы. Возникновение данной проблемы можно проследить в поколениях и обнаружить мутацию в небольшом участке ДНК, непосредственно примыкающем к диверсифицированным генам. Это так называемый центр импринтинга, который каким-то образом указывает на происхождение хромосомы. Импринтинг генов осуществляется с помощью метилирования. 2.2 Клинические проявления и методы диагностики синдромов Прадера-Вилли и Ангельмана 2.2.1 Клинические проявления синдрома Прадера-Вилли Синдром Прадера-Вилли (синдром Прадера-Лабхарта-Вилли-Фанкони, синдром НННО) описан в 1956 году является наиболее частой причиной генетически обусловленного тяжело протекающего ожирения у детей старше 1 года жизни и у взрослых. Встречается с одинаковой частотой среди обоих полов, регистрируется у лиц разных национальностей и рас. Популяционная частота составляет от 1:10 000 до 1:15 000 [5, 6]. Клиническая диагностика СПВ на 1-м году жизни затруднительна в силу отсутствия специфической симптоматики при рождении и быстрой спонтанной ликвидации ранних признаков заболевания после первых месяцев жизни. К этим ранним проявлениям синдрома относятся: - вялое шевеление плода, - слабость сосательного рефлекса вскоре после рождения, - развитие мышечной гипотонии в первые месяцы жизни. Уже на 1-м году жизни могут выявляться различные дизморфии лица и конечностей, гипогонадизм и/или гипогенитализм. Наличие у ребенка первых месяцев жизни указанных симптомов нередко приводит врача к ошибочной диагностике пре- и перинатального поражения ЦНС, инфекционного процесса, так как часты нарушения терморегуляции; различных вариантов наследственных миопатий, врожденного гипотиреоза и других болезней. В существенной степени диагностику СПВ на 1-м году жизни затрудняют спонтанное восстановление сосательного рефлекса в первые месяцы жизни и повышение мышечного тонуса. Вместе с тем, развитие дефицитных заболеваний и необоснованное лечение по поводу другой предполагаемой патологии могут сказаться на последующем онтогенезе ребенка, в частности, его интеллектуальном развитии. Этот факт, несомненно, определяет актуальность ранней диагностики СПВ [5]. В большинстве случаев клиническая диагностика СПВ осуществляется после годовалого возраста ребенка, когда развивается 2-я фаза заболевания, характеризующаяся появлением у больного постоянного чувства голода. У ребенка постепенно и стойко формируется поведение «постоянного поиска пищи». Быстро развивается и прогрессирует ожирение, замедляются процессы роста, появляются умеренные признаки нарушения интеллекта, в дальнейшем нарушаются сроки полового развития и порядок появления вторичных половых признаков. Часто присутствует дневная сонливость, нередко по типу нарколепсии, а также ночные апноэ. В этой связи больные СПВ на протяжении своей жизни подвержены синдрому внезапной смерти. В патогенезе развития 2-й фазы заболевания основную роль играет гипоталамическая дисфункция, в том числе дефицит СТГ (гормона роста). Средняя окончательная длина тела без лечения СТГ у мальчиков составляет 155 см, у девочек — 147 см. Диагностика СПВ до развития 2-й фазы заболевания весьма актуальна. Она создает предпосылки для своевременного формирования у ребенка правильного поведения, в частности, пищевого. Коррекция дефицита СТГ, начатая до 18-месячного возраста ребенка, способствует формированию у него правильного телосложения, препятствует избыточному жироотложению, существенно улучшает развитие мышечной моторики. Последнее время в литературе ведется полемика о целесообразности длительной терапии соматотропином при СПВ. Возможности ранней диагностики СПВ в существенной степени повышаются при использовании диагностических методов, в основе которых лежит балльная оценка присутствующих у больного больших и малых признаков. К большим признакам (каждому присваивается 1 балл) относятся следующие: • характерные лицевые симптомы (долихоцефалия с уменьшением битемпорального диаметра, миндалевидный разрез глаз, небольшой рот с опущенными вниз углами и тонкой верхней губой, страбизм); • задержка нервно-психического развития до 6 лет, умеренное снижение интеллекта и проблемы обучения в школьном возрасте; • проблемы при кормлении в первые месяцы жизни с последующей нормализацией сосания в течение грудного периода; • изменения со стороны половой сферы; • мышечная гипотония центрального генеза в раннем детстве; • прогрессирующее ожирение в возрасте от 1 года до 6 лет. К малым признакам (каждому присваивается 0,5 балла) относятся следующие: • снижение двигательной активности плода и инфантильная летаргия; • нарушения рефракции; • снижение пигментации кожи и волос в сравнении с родителями; • следы «потертостей» кожных покровов; • поперечная ладонная складка; • низкорослость к 15 годам с учетом длины тела других членов семьи; • нарушения сна и апноэ во время сна; • маленькие стопы и/или кисти; • дефекты артикуляции и речи; • густая, вязкая слюна; • поведенческие нарушения [2, 5, 6]. Клинический симптомокомплекс Синдрома Прадера-Вилли включает сахарный диабет или нарушение толерантности к глюкозе. Наличие таких важных диагностических симптомов, как мышечная гипотония (Hypotonia), умственная отсталость (Hypomentia), гипогонадизм (Hypogonadism) и ожирение (Obesity), послужило основанием для одного из наименований синдрома – НННО. Тяжелая мышечная гипотония является наиболее ранним симптомом заболевания, возникает уже во внутриутробный период, что объясняет снижение подвижности плода. В ранний постнатальный период имеют место снижение сухожильных, глотательного и сосательного рефлексов, затрудняющих кормление, дыхательные нарушения, малоподвижность, задержка развития двигательных функций. Со второго полугодия жизни мышечная гипотония заметно уменьшается, однако и у взрослых может сохраняться снижение мышечного тонуса. Появляется полифагия, развивается ожирение. Характерно отложение жира преимущественно в области туловища и проксимальных отделов конечностей, на этом фоне кисти и стопы кажутся диспропорционально маленькими (акромикрия). Акромикрия сочетается с клинодактилией, синдактилией, брахимезофалангией. |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||
Рефераты бесплатно, реферат бесплатно, рефераты на тему, сочинения, курсовые работы, реферат, доклады, рефераты, рефераты скачать, курсовые, дипломы, научные работы и многое другое. |
||
При использовании материалов - ссылка на сайт обязательна. |
||