|
Курсовая работа: Цитогенетическая и молекулярно-цитогенетическая характеристика микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди ДжорджиГипогонадотропный гипогонадизм у лиц мужского пола приобретает клиническую выраженность к пубертатному периоду и сохраняется у взрослых. Его особенности – резкое недоразвитие гениталий, скудное вторичное оволосение, снижение либидо и потенции, атрофия тестикулярной ткани, снижение сперматогенеза. Уже с рождения у мальчиков выявляют двусторонний крипторхизм, маленькую, гладкую мошонку и резкую гипоплазию полового члена, часто с фимозом. У лиц женского пола обнаруживают гипоплазию половых губ, позднее появление вторичных половых признаков, задержки менструаций вплоть до аменореи, инфантилизм матки, бесплодие. Больные обоих полов обычно стерильны. Психомоторное развитие детей замедлено, у большинства больных имеется различной формы умственная отсталость, в редких случаях отмечен нормальный или субнормальный интеллект. Больные, как правило, доброжелательны, безинициативны, плохо контролируют свои эмоции, им свойственна резкая смена настроения [6]. К специфическим черепно-лицевым дизморфиям относятся нерезко выраженная микроцефалия, гипоплазия хрящей ушных раковин, деформация и низкое расположение ушей, сдавленный в височных областях лоб, высокое арковидное небо, гипоплазия нижней челюсти, микродонтия с дефектами эмали и кариесом. Примерно у половины больных наблюдаются гипопигментация кожи, волос и радужки, некоторое повышение фоточувствительности. У 75% детей наблюдается гипопигментация кожи, волос и радужки. Часто диагностируется микроцефалия. Психомоторное развитие отстает от возрастной нормы - коэффициент интеллектуального развития - от 20 до 80 ед. (при норме 85-115 ед.). Речь затруднена, словарный запас уменьшен. Встречаются и другие аномалии: микродонтия, сколиоз, эктропион (выворот века), глаукома. При морфологическом исследовании мозга и ЯМР-томографии могут наблюдаться (примерно в 12% случаев) кисты червя мозжечка, аномалии коры головного мозга. Продолжительность жизни больных может достигать 60 лет и более. Согласно данным литературы, патогенез синдрома Прадера-Вилли до настоящего времени остается малоисследованным. Высказываются предположения, что ожирение у больных обусловлено значительным (более чем в 10 раз) усилением синтеза жира из ацетата и крайне низкими процессами липолиза. Гипогонадизм по гипогонадотропному типу может быть связан с дисфункцией гипоталамуса, преимущественно, в области вентромедиального и вентролатерального ядер. Правильность данной точки зрения подтверждается эффективностью лечения больных фармацевтическими препаратами (кломифен), приводившими к увеличению в плазме содержания лютеинизирующего гормона, тестостерона, нормализации показателей почечной экскреции гонадотропинов, сперматогенеза и появлению вторичных половых признаков [5]. Одним из объяснений гипопигментации кожи, волос и радужки служит снижение активности тирозиназы в волосяных фолликулах и меланоцитах, а также уменьшение пигмента в сетчатке. Обращается внимание на повышенный риск развития лейкемии у больных с синдромом Прадера-Вилли. Исследования выявили снижение репарации ДНК (до 65% по сравнению с 97% у здорового ребенка) в лимфоцитах больных с данной патологией. Не исключено, что низкая репарационная способность ДНК может оказать роковое влияние на развитие злокачественных новообразований у лиц с синдромом Прадера-Вилли [5]. Терапия синдрома Прадера-Вилли окончательно не разработана. По данным литературы, комплекс лечебных мероприятий включает лишь диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины). СПВ должен быть заподозрен у детей до 3-летнего возраста при наличии не менее 5 баллов, а у детей старше 3 лет — 8 баллов, при условии присутствия 4 и более больших признаков. Несмотря на высокую диагностическую чувствительность приведенной шкалы (около 90%), во всех случаях для подтверждения диагноза требуются кариотипирование и молекулярно-генетические исследования 15-й пары хромосом. Специфичность этих исследований достигает 100% . Кроме того, от выявленного типа генетических нарушений зависит генетический прогноз потомства. Дети, страдающие СПВ, должны постоянно находиться под наблюдением педиатра, невролога, психотерапевта, эндокринолога и офтальмолога.
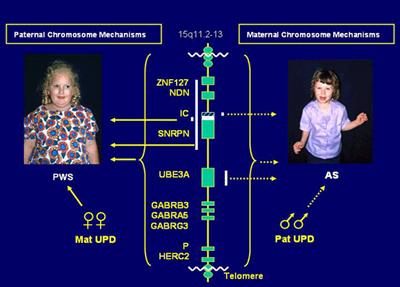
Рис. 4. Девочка с синдромом Прадера-Вилли 2.2.2 Клинические проявления синдрома Ангельмана В 1965 году доктор Гарри Ангельман, английский психиатр, впервые описал трех детей с характеристиками, в настоящее время известными как синдром Ангельмана. Он отметил, что все эти дети имели неуклюжесть, трясущуюся походку, отсутствие речи, излишне смешливы и впадали в припадки. Другие похожие случаи уже были описаны учеными, но эти случаи были особенными, и многие психиатры подтвердили их исключительность. Первые отчеты пришли из Северной Америки в начале 1980-х гг. Синдром Ангельмана — генетическая аномалия. Для него характерны задержка психического развития, нарушения сна, припадки, хаотические движения (особенно рук), частый смех или улыбки. Частота встречаемости, по разным данным, — 1: 10 000-20 000 живорожденных младенцев. (Однако, согласно данным Центра развития человека и отклонений в развитии (университет Вашингтона, США), можно предполагать, что доля людей с синдромом Ангельмана в действительности намного больше статистической.) Синдром Ангельмана обычно не распознается при рождении или в раннем детстве, пока не проявят себя проблемы в развитии, которые не специфичны к этому времени. Родители могут заподозрить диагноз после прочтения о синдроме Ангельмана или после встречи ребенка с такими же признаками. Наиболее распространенный возраст для диагностики – между тремя и семью годами, когда отличительные черты поведения становятся более очевидными. Для синдрома Ангельмана характерны: - В 75 % проблемы с питанием, особенно с грудным вскармливанием, такие младенцы плохо набирают вес; - задержка в развитии навыков общей моторики (умение сидеть, ходить); - задержка речевого развития, неразвитая речь (у всех детей); - дети больше понимают, чем могут сказать или выразить; - дефицит внимания и гиперактивность; - сложности с обучением; - эпилепсия (80% случаев), нарушения выявляются также при электроэнцефалографии; считается, что у детей с синдромом Ангельмана происходит вторичная (симптоматическая) общая эпилепсия; - необычные движения (мелкий тремор, хаотические движения конечностей); - частый смех без повода; - ходьба на негнущихся ногах — из-за этой особенности детей с этим синдромом иногда сравнивали с марионетками; - размер головы меньше среднего, нередко с уплощением затылка; - иногда особые черты лица — широкий рот, зубы с промежутками между ними, выдающийся вперед подбородок, высунутый наружу язык); - нарушения сна; - страбизм (косоглазие) в 40 % случаев; - сколиоз (искривление позвоночника) в 10 % случаев; - повышенная чувствительность к высокой температуре; - бывают сильно увлечены играми с водой [7]. Диагностика: синдром диагностируется путем генетического анализа (15 хромосома), рекомендуемого для новорожденных с пониженным мышечным тонусом (гипотонусом), отставанием в развитии общей моторики и в развитии речи. Возможные методы анализа: процесс флуоресцентной гибридизации in situ, метилирование ДНК в области 15q11-q13, анализ мутации импринтингового центра, анализ прямой мутации гена UBE3A. Существует небольшая группа людей, у которых результаты всех вышеописанных анализов в норме, однако наблюдаются все внешние проявления синдрома Ангельмана. Наука пока не дает ответ на вопрос, как это возможно. Лечение: синдром Ангельмана является врожденной генетической аномалией и, следовательно, не может быть излечен. Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии). Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом. Нарушения сна корректируются назначением легких снотворных. Д-р Вагстафф (США) считает, что назначение 0.3 мг мелатонина за 30 минут-1 час перед сном улучшает сон пациентов с синдромом Ангельмана. А нарушения стула регулируются назначением легких слабительных. Приступы лечатся так же, как эпилепсия. Дети с синдромом Ангельмана часто испытывают больше одного типа приступов. Д-р Чарльз Вильямс (Гейнсвилл, Флорида), работающий в основном с аутичными детьми, отмечает общие для аутичных детей и детей с синдромом Ангельмана особенности поведения: заметная аутостимуляция, импульсивность, навязчивые, повторяющиеся движения, интерес к неуместным предметам, а также сложность в общении с другими людьми. Врачи США показывают, что для аутичных детей внутривенные инъекции гормона секретина успешно уменьшают проявления нежелательного поведения и обеспечивают хороший уровень общительности и коммуникативных навыков; возможно, медицина придет к использованию секретина для коррекции поведения детей с синдромом Ангельмана [2, 7]. Риски: оценка риска повторного рождения ребенка с синдромом Ангельмана у тех же родителей очень сложна, необходима консультация врача-генетика. Считается, что обычная делеция является спонтанной, риск повтора меньше 1 %. В случае молекулярной микроделеции в 15q11-q13, если она наблюдается и у матери, риск теоретически до 50 %. Мутации внутри гена UBE3A могут быть случайными и неунаследованными, в этой ситуации риск повтора <1%; однако эти мутации можно унаследовать от нормальной матери, и тогда теоретический риск 50%. Партеногенетическая дисомия 15-ой пары – случайная ситуация; риск повтора <1%. Есть несколько людей с AS с необычными преобразованиями хромосомы 15, включая область 15q11-q13; в этих случаях оценка риска повтора зависит от хромосомных нарушений у родителей [7]. Перспективы развития: дети с синдромом Ангельмана понимают намного больше, чем могут сказать. В некоторых случаях у них вообще нет речи; описаны дети со словарем 5-10 слов. При этом дети/люди с синдромом Ангельмана любят общаться с людьми, играть. Рекомендуется обучать таких детей языку жестов. Занятия с раннего возраста по специальным программам, направленные на развитие навыков мелкой и общей моторики, в ряде случаев дают хорошие результаты. Перспективы развития зависят от степени пораженности хромосомы. Некоторые люди с синдромом Ангельмана способны освоить навыки самообслуживания и речь на примитивном уровне (обычно причиной синдрома в этом случае стала мутация), некоторые никогда не смогут ходить и говорить (это обычно происходит в случае делеции части хромосомы). С возрастом, как правило, симптомы гиперактивности и нарушения сна смягчаются. У девочек с синдромом Ангельмана в период полового созревания могут участиться припадки. Большинство людей с синдромом Ангельмана способны контролировать экскреторные функции (мочеиспускание и дефекацию) днем, некоторые — и ночью. Некоторые люди с синдромом Ангельмана способны есть ножом и вилкой, одеваться самостоятельно в случае отсутствия на одежде пуговиц, «молний». Во взрослом возрасте может появиться ожирение и ухудшиться ситуация со сколиозом. Менструации, половое созревание индивидов с синдромом Ангельмана происходит в обычные сроки. Описан один случай беременности женщины с синдромом Ангельмана: она родила девочку с таким же диагнозом [6, 7, 8]. Таким образом, несмотря на повреждение при синдромах Прадера - Вилли и Ангельмана одного и того же локуса хромосомы 15, клинические проявления обеих болезней резко противоположны. 2.3 Связь синдромов Прадера-Вилли и Ангельмана Установлено, что формирование синдрома Прадера-Вилли и синдрома Ангельмана связано с геномным импринтингом [4]. В большинстве случаев синдрома Прадера-Вилли (70%) микроделеции района 15q11-q13, обнаруживаемые у пациентов, имеют отцовское происхождение. Примерно в 30% случаев у пациентов обнаруживаются только материнские аллели (однородительская материнская дисомия). Эта своеобразная «гомозиготизация» происходит в результате неравного кроссинговера или соматической рекомбинации по механизму конверсии генов. Синдром Ангельмана является как бы зеркальным отражением синдрома Прадера-Вилли. Делеции, захватывающие примерно тот же район хромосомы 15, что и при синдроме Прадера-Вилли, имеют материнское происхождение, а однородительская дисомия - отцовское. Установлено, что в перицентромерном участке хромосомы 15 имеется тандемный инвертированный повтор MN7 [4], который, по-видимому, определяет высокую мутабельность района 15q11.2. Пациенты с интерстициальной дупликацией проксимального района 15q могут иметь задержку развития без признаков дисморфогенеза. С целью определения степени выраженности патологии пациенты с дупликацией родительского происхождения критического региона 15q были разделены на 3 группы. К первой группе были отнесены больные с эухроматиновым вариантом дупликации без захвата локуса 15q11-q13. Вторая группа включала пациентов с дупликацией 15q11-q13 материнского происхождения, у которых имели место трудности обучения и речи, признаки аутизма или атипичный аутизм без дисморфологических черт. Третья группа включала пациентов с дупликациями отцовского происхождения без очевидного клинического фенотипа [4]. У одного больного, имевшего дупликацию 15q отцовского происхождения с вовлечением локуса 15q11-q13, были выявлены задержка развития речи и аномалия мозга. Инвертированная дупликация inv dup 15 с вовлечением локуса 15q11-q13 была обнаружена у детей с задержкой развития, умственной отсталостью от умеренной до тяжелой, эпилепсией, манифестировавшей в возрасте от 4 до 8 лет, генерализованной гипотонией и аутистическим поведением, но без дисморфологических черт или с 1-4 малыми аномалиями развития [4]. К настоящему времени описано два цитогенетических типа инвертированной дупликации хромосомы 15. Первый тип включает метацентрик или субметацентрик и гетерохроматиновую хромосому, меньшую или равную хромосомам G группы, - dic(15)(q11). Большинство детей с данной аномалией имеют нормальный фенотип. Второй тип включает хромосому большего размера, чем хромосомы группы G, и имеет 15q эухроматиновый участок. Данный участок включает 15q11-q13 регион и его цитогенетическое описание - dic(15)(q12 или q13). Этот дицентрик получен из двух соответственных материнских хромосом в мейозе и обычно связан с большим возрастом матери и аномальным фенотипом, описанным выше. Таким образом, при подозрении на данную хромосомную аномалию необходимо сочетать стандартный цитогенетический анализ с FISH-анализом. Задержка развития легкой степени с Прадер-Вилли-подобным фенотипом описана у пациентов с однородительской дисомией хромосомы 14 материнского происхождения. Однородительская дисомия хромосомы 14 отцовского происхождения также может быть ассоциирована с различными аномалиями. Однородительская дисомия хромосомы 14 является, таким образом, одной из возможных причин задержки развития и должна выявляться в подозрительных случаях методом ДНК-анализа. Это один из путей улучшения диагностики умственной отсталости. С. Williams и соавторы привлекли внимание к состояниям, по своим фенотипическим проявлениям напоминающим синдром Ангельмана. Данные наследственные формы могут быть вызваны как хромосомными аберрациями, так и генными мутациями. Могут отмечаться терминальные делеции 22q13.3, реже микродупликации 15q11-13, интерстициальные делеции 2q21-23, 17q23.3, 4q. По мнению автора, все эти хромосомные аномалии должны быть проанализированы при обследовании пациентов с фенотипическими особенностями, напоминающими синдром Ангельмана. Одним из синдромов, требующих дифференциальной диагностики с синдромом Ангельмана, является синдром Ретта, особенно учитывая, что многие пациенты с данным состоянием умирают в младенчестве или раннем детстве. Отсутствием речи, атаксией, эпилептическими приступами и поведенческими особенностями, характерными для синдрома Ангельмана, может проявляться дефицит метилен-тетрагидрофолат-редуктазы. Глубокая умственная отсталость и протрузия языка могут быть проявлениями АТR-Х синдрома (Х-сцепленный рецессивный синдром альфа-талассемии).А. Battaglia и соавторы приводят алгоритм полного генетического исследования пациентов с подозрением на синдром Ангельмана [6]. Сначала необходимо исключить синдром Ангельмана ДНК-анализом. В случае отрицательного результата провести цитогенетический анализ с высоким разрешением полос, затем субтеломерный анализ (чтобы исключить делецию 22q13.3), затем методом сравнительной геномной гибридизации исключить синдромы, сопровождающиеся интерстициальными делециями, и наконец, использовать ДНК-анализ для выявления мутаций в гене МЕСР2. Если все анализы дали отрицательный результат, необходимо провести полное цитогенетическое и молекулярно-генетическое исследования региона 15q11-q13, исключить дефицит метилен-тетрагидрофолат-редуктазы и мутации в гене АТR-Х [6]. Те же авторы приводят алгоритм обследования больных с мышечной гипотонией и Прадер-Вилли-подобным фенотипом. Этим пациентам необходимо провести ДНК-исследование районов метилирования, исключить синдром FRA-Х, затем осуществить ДНК- анализ на однородительскую дисомию хромосомы 14 и в случае отрицательных результатов провести исследование для выявления МЕСР2 мутаций. Таким образом, чтобы провести полный генетический анализ при подозрении на синдромы Ангельмана или Прадера-Вилли, в некоторых случаях необходимо применить до 8 различных цитогенетических, молекулярно-цитогенетических, молекулярных методов. Пациенты с подозрением на указанные синдромы являются примером многостороннего и целенаправленного исследования, необходимого для правильной постановки диагноза больным [6].
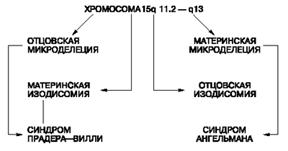
Рис. 5. Возможные механизмы развития синдромов Прадера-Вилли и Ангельмана В заключение следует еще раз подчеркнуть, что более широкое комплексное применение цитогенетических, молекулярно-цитогенетических методов лабораторного анализа в сочетании с грамотным клиническим обследованием позволит решить проблему диагностики изолированной умственной отсталости или умственной отсталости при наследственных болезнях и синдромах у детей. Это в свою очередь поможет более эффективно осуществлять помощь членам наследственно отягощенной семьи в решении вопросов прогноза потомства и снизить частоту рождения детей с генетическими патологиями. ГЛАВА 3. СИНДРОМ ДИ ДЖОРДЖИ (ДИ ГЕОРГА) Синдром Ди Джорджи (Ди Георга, СДД) - изолированный Т-клеточный иммунодефицит. Характеризуется триадой ведущих клинических проявлений: гипоплазия тимуса и/или паращитовидных желез и врожденным пороком сердца. Впервые описан в 1965 году. Поражаются чаще девочки [9, 10, 11]. Наследуется по аутосомно - рецессивному типу [11]. Установлена генетическая характеристика этого врожденного порока развития, в части случаев это аутосомный генетический дефект делеции 22q11.2. В основе СДД лежит порок развития третьего-четвертого глоточных карманов, возникающий между шестой и десятой неделями гестации, приводящий к агенезии или дисгенезии паращитовидных желез и тимуса. Вовлечение первого и второго жаберных карманов приводит к пороку развития лицевых структур, а заинтересованность пятого кармана проявляется широким спектром врожденных пороков сердца с частым вовлечением дуги аорты [9]. Клиническая характеристика: у большинства больных отмечаются диспластические черты лица. Наиболее характерны диспластичные ушные раковины, гипертелоризм, широкая переносица, "рыбий рот", антимонголоидный разрез глаз. У части детей наблюдаются и более грубые аномалии, такие как микрогнатия и незаращение твердого и мягкого неба. Гипокальциемия различной степени тяжести и отсутствие тени вилочковой железы при рентгенографии грудной клетки относятся к частым проявлениям. Гипокальциемические судороги обычно возникают с первых дней жизни. У всех больных отмечается задержка умственного развития. Врожденные пороки сердца и магистральных сосудов также относятся к наиболее характерным и тяжелым признакам заболевания [9]. Тимус и паращитовидные железы образуются из третьего и частично четвертого карманов. Паращитовидные железы участвуют в регуляции уровня кальция в крови. Отсутствие данных желез приводит к судорогам вследствие гипокальциемии. Появление судорог у младенца в течение 48 часов после рождения свидетельствует о том, что у ребенка врожденная аплазия тимуса и дефицит Т-клеток. С данным синдромом связаны некоторые другие пороки развития, в том числе расщелина нёба (волчья пасть), щели в носу, низкопосаженные уши и неправильное формирование крупных кровеносных сосудов и сердца. Дети с пороками развития сердечно-сосудистой системы, как правило, умирают. Одно время считали, что этот синдром вызван нарушением нормального эмбрионального развития, которое не обусловлено наследственностью, поскольку не наблюдается семейной предрасположенности к заболеванию и оба пола поражаются одинаково. Впоследствии, однако, было установлено, что многие случаи заболевания связаны со структурными дефектами в 22-й хромосоме [9]. У детей, страдающих синдромом Ди Джорджи, либо совсем нет тимуса, либо он недоразвит, что встречается чаще. Такие дети чрезвычайно восприимчивы к вирусным, бактериальным и грибковым инфекциям и склонны к развитию таких опасных для жизни заболеваний, как цитомегаловирусная инфекция. Иногда в крови ребенка присутствует некоторое количество Т-клеток; такие дети способны сопротивляться инфекциям, при этом с возрастом число Т-клеток постепенно увеличивается [11]. Одна из форм нарушения иммунитета. Проявляется с рождения. Основные симптомы — тетания, обусловленная гипопаратиреозом; повышенная склонность к инфекциям, особенно вирусным и грибковым; аномалии развития костной системы, полости рта, сосудов, внутренних органов. Из инфекционных болезней чаще возникают бронхиты, пневмонии, диарея. Дети отстают и физическом развитии. Болезнь прогрессирует и без лечения к двум годам наступает летальный исход. При исследовании крови определяется низкий уровень, содержании кальции и высокий уровень фосфора, указывающие на паратиреендную недостаточность. Число лимфоцитов в периферической крови обычно нормальное, уровень иммуноглобулинов основных классов также в пределах нормы, но специфический иммунный ответ на введенный антиген снижен. Снижены реакции клеточного типа. Не развиваются и ослаблены реакции замедленной ин нечувствительности. Стимулированный лимфоузел обнаруживает избыточное число фолликулов и плазматических клеток. Наблюдаются уменьшение числа лимфоцитов в паракортикальных тимусзависимых зонах и белой пульпе селезенки, аплазия тимуса, гипоплазия паратиреоидных желез или аплазия их. Аплазия тимуса ведет к нарушению синтеза полноценных иммуноглобулинов. [12]. Иммунологический спектр: количественные показатели Т-клеток варьируют от нормы до глубокой депрессии. Характерна диссоциация между сниженными уровнями Т- и NK-клеток и повышенным содержанием В-лимфоцитов. Характерны нормальные или повышенные уровни антител [9]. Показатели гуморального иммунитета у больных с синдромом Ди Джорджа полностью нормальны. Концентрация иммуноглобулинов всех классов нормальная. У некоторых больных повышена концентрация lgE, что, возможно, связано с отсутствием Т-супрессоров. У части больных не удается получить антитела в ответ на иммунизацию [11]. 1. Обязательные лабораторные исследования: - Анализ крови клинический4 - Тромбоциты 2 - Определение группы крови и резус фактора1 - Анализ мочи2 - Определение уровня кальция в крови2 - Бактериологические исследования содержимого из очагов с определением чувствительности к антибиотикам1 - Общий белок и белковые фракции1 - Трансаминазы АсАТ, АлАТ1 - Маркеры вирусов гепатита В и С в крови1 - С-реактивный белок1 - Сывороточные иммуноглобулины А, М, G1 - Субпопуляции лимфоцитов2 - Концентрация общего IgE в сыворотке1 2. Дополнительные лабораторные исследования: - Определение уровня паратгормона в плазме1 - Вирусные маркеры (антитела к вирусу Эпштейна-Барр)1 3. Обязательные инструментальные исследования: Рентгенография органов грудной клетки УЗИ сердца ЭКГ 4. Дополнительные инструментальные исследования: УЗИ органов брюшной полости - 1. 5. Консультации специалистов (по показаниям): Эндокринолога - 1; Кардиохирурга - 1; Отоларинголога - 1; Окулиста - 1. [7] Характеристика лечебных мероприятий: иммунная недостаточность, за редким исключением, не определяет прогноз и спектр ведущих клинических проявлений при СДД. В большинстве случаев, если пациент переживает 6-месячный возраст, наблюдается постепенное спонтанное восстановление Т-клеточного иммунитета. Коррекция Т-клеточных нарушений при СДД может быть достигнута трансплантацией фетального тимуса. При наличии тяжелых пороков, в основном определяющих прогноз для жизни, пересадка тимуса считается недостаточно обоснованной. Лечение пороков сердца ведется по стандартам, принятым в кардиологии, а недостаточности паращитовидных желез - по стандартам эндокринологических отделений [9]. Для лечения синдрома Ди Джорджи с успехом применяют пересадку костного мозга. Кроме того, достигнуты положительные результаты при пересадке эмбрионального тимуса больным с тяжелой степенью поражения [11]. Лечение: трансплантация или имплантация ткани тимуса. Введение тимозина (экспериментально). Противосудорожная терапия, введение препаратов кальция [11]. Длительность стационарного лечения определяется характером и тяжестью клинических проявлений и инфекционных осложнений и варьирует от 3 недель до 3 месяцев [9]. Требования к результатам лечения - купирование клинических проявлений (осложнений) СДД [9, 10]. ВЫВОДЫ В представленной работе были рассмотрены микроделяционные синдромы: Прадера-Вилли, Ангнльма и Ди Джорджи. Описаны их клинические и генетические проявления, возможные риски. И представлены медицинские методы купирования их проявлений, улучшения состояния больных. На сегодняшний день синдромы Прадера-Вилли и Ангельмана служат общепринятой моделью для изучения новых в клинической генетике и сложных явлений - геномного импринтинга и унипарентальной дисомии. Установлено, что синдром Прадера-Вилли может быть обусловлен двумя основными механизмами. Первый из них - микроделеция хромосомы 15 (15q11.2-q13), которая всегда отцовского происхождения. Второй - материнская изодисомия, т.е. когда обе хромосомы 15 получены от матери. Развитие синдрома Ангельмана, наоборот, связано с микроделецией того же участка хромосомы 15, но материнского происхождения, или отцовской изодисомией. Большинство (около 70%) случаев синдрома Прадера - Вилли обусловлено микроделецией, остальные - дисомией. При этом обращает на себя внимание отсутствие клинических различий между больными с микроделецией и изодисомией. Для синдрома Ди Джорджи установлено, что он вызывается аутосомным генетическим дефектом делеции 22q11.2. Цитогенетические проявления данного синдрома на сегодня еще не достаточно изучены. Учеными ведется активная работа по решению этой проблемы. СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ 1. Гинтер Е.К. Медицинская генетика: Учебник. – М.: Медицина, 2003. – 448с. 2. Ридли М. Геном. Автобиография вида в 23 главах. – М.: Эксмо, 2008. – 432с. 3. Денисенкова Е.В., Петрин А.Н., Байков А.Д. Цитогенетические и молекулярно-цитогенетические аспекты диагностики заболеваний, сопровождающихся умственной отсталостью, у детей // Издательство «Медиа Сфера», 2005. Режим доступа: http://www.mediasphera.ru/journals/detail/206/2988/ 4. Сапиенца К. Геномный импринтинг // В мире науки. (Scientific American. Издание на русском языке). - 1990. - №12. - стр.14-20. Режим доступа: http://www.evolbiol.ru/gen.html 5. Дуліпа О.Г. Синдром Прадера-Віллі (Prader-Willi Syndrome). Режим доступа: http://www.ibis-birthdefects.org/start/ ... prader.htm 6. Казанцева Л.З., Новиков П.В., Семячкина А.Н., Николаева Е.А., Курбатов М.Б., Добрыкина Э.В. Московский НИИ педиатрии и детской хирургии Минздрава РФ. Синдром Прадера - Вилли у детей: новое в этиологии, патогенезе и лечении. Режим доступа: http://nature.web.ru/db/msg.html?mid=1174772&uri=index2.html 7. Миронов М.Б., Мухин К.Ю., Кузина Н.Ю., Боровиков К.С., Гоева И.А., Красильщикова Т.М., Коркин А.В., Петрухин А.С. Синдром Ангельмана. Клинический случай. Режим доступа: http://www.epileptologist.ru/pub/Angelman.html 8. Белозеров Ю.В., Леонтьева И.В., Школьникова М. А., Страхова О.С., Себелева И.А., Макаров Л.М., Давыдкин В.В., Динов Б.А. Наследственные болезни сердца у детей // Мир науки и культуры, 2000-2009. Режим доступа: http://nature.web.ru/db/msg.html?mid=1166264&s=111400060 9. Соколов В.Н. Гипопаратериоз // Наука и практика, 2009. Режим доступа: http://www.chtfoms.ru/index.php?option=com_content&task=view&id=26467&Itemid=24 10. Ярцев М.Н., Яковлева К.П., Плахтиенко М.В. ГНЦ Института иммунологии ФМБА России, Москва Иммунодефицитные состояния у детей // Издательство Media Medica, 2000. Режим доступа: http://old.consilium-medicum.com/media/pediatr/06_01/4.shtml 11. Вилочковой железы аплазия врожденная (Ди Джорджа синдром). Режим доступа: http://nasledbolezn.org.ua/obmen-veshestv/didjordja-sindrom.html ПРИЛОЖЕНИЕ
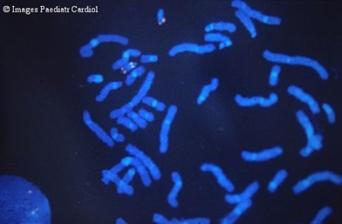
Рис. 1. 15 хромосома
Рис. 2. Делеция 15 q11 – 13.1
Рис. 3. Дети с синдромом Прадера-Вилли
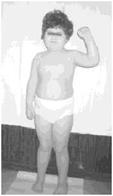
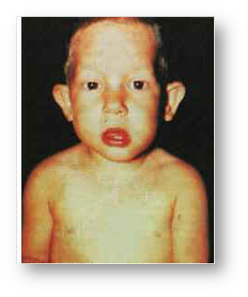
Рис. 4. Мальчик с синдромом Прадера-Вилли
Рис. 5. Синдромы Прадера-Вилли и Ангельмана
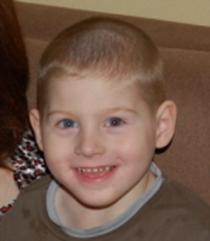
Рис. 6. Мальчик с синдромом Ангельмана
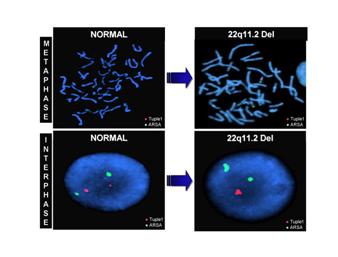
Рис. 7. Делеция 22q11.2 – синдром Ди Джорджи
Рис. 8. Делеция 22q11.2 – синдром Ди Джорджи
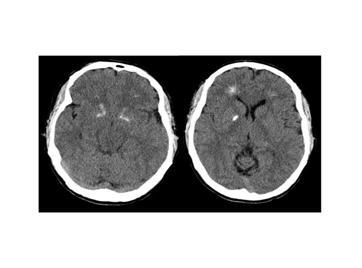
Рис. 9 Компьютерная томограмма мозга пациента с делецией
Рис. 10. Синдром Ди Джорджи
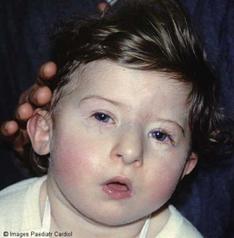
Рис. 11. Девочка с синдромом Ди Джорджи
Рис. 12 Мальчик с синдромом Ди Джорджи |
|||||||||||||||||||
 |
||
| НОВОСТИ |  |
|
|
||
| ВХОД | |
|
|
|||||
Рефераты бесплатно, реферат бесплатно, рефераты на тему, сочинения, курсовые работы, реферат, доклады, рефераты, рефераты скачать, курсовые, дипломы, научные работы и многое другое. |
||
При использовании материалов - ссылка на сайт обязательна. |
||